

Atherosclerosis

Atherosclerosis is a chronic, diffuse disease of large arteries of unknown direct cause but clearly associated with diet and tobacco. In “Westernized” societies the disease begins in childhood and progresses inexorably unless lifestyle is changed. One prevalent hypothesis is that it is caused by damage to the endothelium, the single-cell thick layer lining all blood vessels and the largest organ in the body. Until about 20 years ago the endothelium was thought to be a passive organ whose only job was to prevent clotting of blood on the inside of blood vessels. But since the Nobel Prize winning research of Furchgott, Ignarro and Murad we know that endothelium is a very active organ whose normal function is to keep the arterial wall healthy but which is also very sensitive to substances in the blood and the flow of blood itself. Healthy endothelium releases nitric oxide, a gas that keeps the arterial wall healthy but that is rapidly inactivated by blood. A number of factors can cause endothelial dysfunction, the most important being, diet, tobacco, and inactivity. By as yet unknown mechanisms, possibly related to the production of modified LDL (low density lipoprotein) in the arterial wall, endothelial dysfunction leads to the build up of atherosclerotic plaque. Most importantly, endothelial dysfunction and plaque are reversible if those damaging factors are removed.

Endothelium-derived nitric oxide (EDNO) synthesis and action. In endothelial cells, a constitutive membrane-associated NO synthase (eNOS) catalyzes the conversion of the amino acid L-arginine to NO and L-citrulline. eNOS cofactors include FAD (flavin adenine dinucleotide), NADPH (nicotinamide adenine dinucleotide phosphate), TH4 (tetrahydrobiopterin), and calcium. Endothelial synthesis of NO is tightly controlled and linked to changes in ionized calcium concentration. Several agonists, including acetylcholine, bradykinin, substance P, and platelet-derived serotonin, act on specific membrane receptors that trigger cytosolic calcium release and eNOS activation. Increased shear stress from enhanced blood flow also serves as an important stimulus for NO production. Nitric oxide modulates basal vascular tone, and exerts a dilator effect on vascular smooth muscle through activation of soluble guanylyl cyclase and consequent increase in intracellular cyclic 3',5'-guanosine monophosphate (cGMP), which also mediates NO-dependent inhibition of platelet activation. Individuals with coronary risk factors or atherosclerosis demonstrate impaired shear stress- and agonist-induced endothelium-dependent vasodilation. Other actions of NO include inhibition of monocyte adhesion and smooth muscle cell proliferation. GTP (guanosine 5'-triphosphate).

The response-to-injury hypothesis of atherosclerosis. In response to mechanical injury or exposure to atherogenic stimuli, such as oxidized LDL, diabetes mellitus, severe hyperhomocystinemia, and cigarette smoking, endothelial cells express adhesion molecules and elaborate growth factors that lead to recruitment of leukocytes in an inflammatory response to injury. Leukocytes adhere and migrate into the vessel wall, localize subendothelially, and develop into lipid-laden macrophages (foam cells). Foam cells, in turn, release growth factors and cytokines that promote recruitment of smooth muscle cells and stimulate neointimal proliferation, continue to accumulate lipid, and support endothelial cell dysfunction. Collectively, these events promote the development of a lipid-rich atheromatous lesion. Subsequent denudation of the endothelium exposes circulating platelets and coagulants to the underlying matrix, thereby initiating thrombosis, and triggering a cascade of events leading to a fibroproliferative lesion and luminal narrowing.

Note that as the plaque grows the lumen of the artery remains a constant diameter because the wall of the artery dilates to accomodate the plaque. So just doing an angiogram that only measures lumen diameter will not detect even large plaques that could rupture and cause a heart attack. Angiograms can be very deceptive and have no prognostic value.

Intimal surface of right coronary artery from a 32-year-old man. Atherosclerotic lesions are advanced when the accumulation of lipid, cells, and matrix are associated with disorganization, repair, and thickening of the intima and deformity of the arterial wall. Type IV lesions, also called atheromas, are advanced lesions in which extracellular lipid occupies a well-defined and extensive area in the intima, called the lipid core, that develops by coalescence of smaller isolated pools of extracellular lipid. When seen in younger people, atheromas occur in areas of previous adaptive intimal thickening. Although atheromas are raised lesions, they do not necessarily result in significant luminal occlusion. Microscopically, the extracellular lipid in atheromas displaces and replaces smooth muscle cells and extracellular matrix.

A cross-section of coronary artery that has significant luminal obstruction by an atheroma. A lipid-rich core can be seen in the center of the lesion.

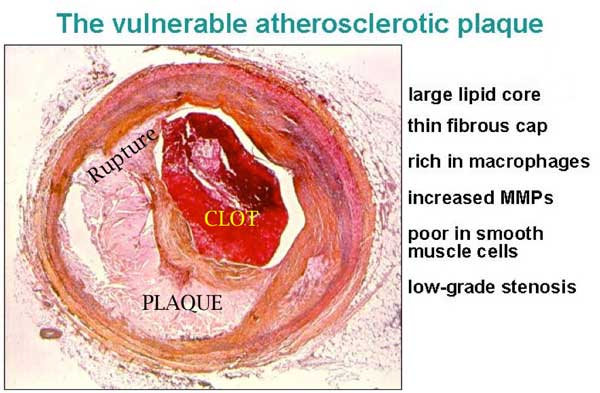
Rupture of a plaque. Note that the endothelium is broken exposing the underlying tissue to the blood and starting the clotting cascade, in the same way blood clots when blood vessels in the skin are cut. Plaque rupture is cut inside an artery.

Using the technique of intravascular ultrasound (IVUS) in hearts removed for transplantation from accident victiims of all ages in the US, investigators at the Cleveland Clinic found advanced atheroscerotic plaque in one sixth of teenagers. By age 50, 85% of hearts had advanced plaque. None of these subjects had symptoms or had yet had a heart attack but were clearly at risk for one because the plaque was buried in the arterial wall which had undergone compensatory dilation. Obviously prevention of atherosclerosis must begin in childhood. Unless one is foolish enough to believe that statins should be given to every child in the Western world, prevention means adoption of a non-atherogenic lifestyle from weaning.
Fortunately, as plaque builds up the artery tends to dilate in an attempt to keep the lumen open. This phenomenon is call compensatory dilation.
Blockage of arteries by atherosclerosis usually first causes symptoms related to heart function because the coronary arteries that deliver blood to the heart muscle are only about 2 mm in diameter. The heart muscle (myocardium) extracts more oxygen from its blood supply than any other organ in the body and cannot tolerate any reduction in blood flow.
Angiography outlines the lumen of the artery and, because the wall is not visualized, shows only a small fraction of the disease, the “tip of the iceberg”.
Heart attacks are caused by rupture of atherosclerotic plaques, formation of a clot and rapid occlusion of a coronary artery. When the endotheilum, which normally provides a non-thrombogenic surface is broken, blood is exposed to tissue factors in the wall of the artery which promote clotting. The same happens with any trauma. Blot clots when exposed to tissue factors outside of the endothelium.
Sudden death is usually caused by a heart attack. Heart muscle, as well as contracting, also conducts electical impulses like a nerve. When a piece of heart muscle dies the normal smooth flow of conduction is disturbed and can result in a totally disorganized kind of electrical activity called ventricular fibrillation and an absence of coordiinated pump function.
Paradoxically, a heart attack or sudden death is much more likely to be caused by plaque rupture in regions of the diseased artery which are not very constricted and not even seen on an angiogram, early unstable plaque. These procedures will not prolong life or prevent a heart attack as proven by the COURAGE trial. Angioplasty (balloon dilation) or graft bypass of the constriction may relieve angina temporarily but leaves untouched the great majority of the disease. Likewise, opening a completed occluded artery supplying the dead piece of heart muscle more than three days after a heart attack does not prevent more heart attacks or prolong life and may even be dangerous, as shown by the OAT study.
Many cardiologists and cardiovascular surgeons still labour until the profitable but mistaken notion that the larger, obstructive plaques are more likely to cause a heart attack and recommend “revascularizaton” solely on that basis, even in the absence of symptoms. This futile treatment costs medical systems $many billions.
Plaque rupture can be prevented and the plaque stabilized by regression of the fatty deposits in the plaque. Regression can only be accomplished by adoption of a low-fat, mostly vegetarian diet, exercise and smoking cessation. Except in high, potentially toxic doses, cholesterol lowering drugs will only slow progression of atherosclerosis not reverse it.

Pourquoi certaines plaques d’athérosclérose sont mortelles ? | Vulgariz - vulgarisation scientifique said
[…] Kuriakose, David Ron, Ira Tabas Journal de publication : Cell Metabolism — Illustrations : Panaceia or Hygeia Liens externes: Articles similaires:Les hommes et les femmes ne sont pas égaux devant la […]
tammikettu said
This was an excellent description but one thing in the very end is probably wrong:”Regression can only be accomplished by adoption of a low-fat, mostly vegetarian diet …”
The latest studies have pointed out that e.g. saturated fat has no correlation to CVD. Also the increased amount of carbohydrates to replace the fat may be a reason for CVD, obesity and diabetes. So low-fat cannot be the cure.
Colin Rose said
The issue is not saturated vs. unsaturated fat. It’s total fat. Refined fat of any sort has no significant nutrients other than naked calories. The diets shown to reverse atherosclerosis have all been low-fat.
No one is recommending fat be replaced by refined carbohydrates. Unrefined carbohydrates have repeatedly been shown to be beneficial for a number of diseases of lifestyle.
See one of my other blogs for details.
olson012 said
I would like to use the picture of atherosclerotic plaque in a publication. Please send contact for this.
Colin Rose said
I can’t remember where I got those illustrations. They are from various web sites and CME material and I have modified some to correct mistakes and improve the labelling.
iagorc said
I would like to know what´s your opinion on coronary calcium scans, the SHAPE guidelines, etc…
Colin Rose said
There are a number of invasive and non-invasive imaging tests for detection of coronary atherosclerosis in asymptomatic people. None have ever been shown in any RCT to prevent heart attacks or prolong life. At any rate, we know that by middle age the vast majority of the populations of “developed” countries have advanced coronary atherosclerosis, a preventable disease.
SHAPE was founded by Morteza Naghavi who is an agent for CardioNexus Corp. which sells a Panasonic machine for measuring CIMT. Funding for SHAPE comes from Panasonic. It is just a lobby for imaging machine manufacturers. SHAPE’s recommendations are not scientifically validated and should be ignored.
The problem is that there is no drug or operation proven to prevent heart attacks even if one knows one’s CIMT or calcium score or IVUS plaque volume. Heart attacks are caused by rupture of an early unstable coronary plaque and there is, to date, no way or predicting which one is in danger of rupturing.
There are at least three good studies showing that statins have no effect on progression of calcium scores in primary prevention.
Atherosclerosis is caused by junk food and/or tobacco addiction. If these addictions didn’t exist there would be no need for any imaging test for early atherosclerosis.
Dan Boyle said
Dr Rose,
I visited you yesterday, and I’m glad you were able to give me access to this information along with the explanation you provided. Thank you. My question may help others. What impact does stress have on the coronary system? I forgot to mention to you that I’m under quite a lot of stress.
Thanks,
Dan Boyle
Colin Rose said
There is evidence both in lower primate and human epidemiological studies that social status in a group is correlated with atherosclerosis but we don’t know why there is this relationship. Are higher ranking individuals with more responsibility more or less stressed than lower ranking individuals who have to make less decisions? At any rate this effect is minor compared to diet and exercise. The main effect of stress in our society is that it makes people less attentive to their lifestyle and, for example, to grab junk food on the run instead of sitting down to a healthy meal or to feel they deserve some junk food after a stressful day at the office.